We know this is not the topic headline to end all topic headlines.
It is boring to say the least, but hopefully you will nevertheless find this article helpful, since it gives you some insight into the FDA world of approving clinical trials.
The FDA Draft Guidance
But “what lies beneath” is The Food and Drug Administration (FDA or Agency) having announced December 19th. 2019, an availability of a draft guidance for industry entitled “Demonstrating Substantial Evidence of Effectiveness for Human Drug and Biological Products.”
Also known as Docket (FDA-2019-D-4964).
This was to complement and expand on the 1998 guidance entitled “Providing Clinical Evidence of Effectiveness for Human Drug and Biological Products”.
The new final Guidance, has been long underway and will hopefully be coming soon and it will hopefully be ground-breaking, and pave the way for allowing clinical trials using externals control arms (ECAs), and maybe a different way of thinking about statistical analysis, including p value.
A Guide to the Guidance
A Guide has been made to guide you through this complex world of setting up trial designs, trial endpoints and more, and which actually contains - if not answers then at least a lot of informations - to a lot of questions and even accusations and claims revolving around NWBO’s trial design and endpoints.
The guide is called “FDA’s Evolving Interpretation of How Sponsors Can Demonstrate Substantial Evidence of Effectiveness for Human Drug and Biological Products”.
FDA made it possible to submit comments from January 2020 to the guidance while still in draft, and stated
Submit either electronic or written comments on the draft guidance by February 18, 2020 to ensure that the Agency considers your comment on this draft guidance before it begins work on the final version of the guidance.
Linda Powers submitted her comments
Comments from flipper44:
Since this is before the trial was unblinded to LP, her comments regarding external control arms and allowing more flexible analysis regarding subtypes should be viewed as a derisking statement, as should her comment about more flexible p value rules.
Since that time, in September 2020, the phase iii trial was allowed to derisk their trial by modifying its primary endpoint to utilize external control arms. That trial’s new endpoints also contemplate adding recurrent GBM as an additional potential treatment indication. The trial also kept/converted the original primary endpoints as/into secondary endpoints. That allows a hybrid trial, that compares the treatment group to both external control AND placebo control. This design is often used to strengthen the confidence and power of results, whilst being triggered by such things as an ethical dilemma of not placing more placebos into a trial, where the placebo + standard of care is futile. The hybrid technique is also used to accommodate trials with crossovers, because trials with crossovers are otherwise only ultimately comparing an experimental group where the treatment was provided early versus an experimental group where the treatment was given later in time. That said, Dr. Liau has since stated, based upon her non-phase iii trial practice experience, in approximately April 2019, that it is important people get DCVax-l early.
It should also be noted that the FDA guidance LP commented on is not required in order to allow external control arms in orphan designated trials/indications. In fact, this method has been used since 2001.
Another follow up point is the redefinition of GBM occurred since LP made those comments, which now excludes idh mutant tumors from the GBM definition. It just so happens, DCVax-l is considered far more likely to be effective against the remaining subtypes of GBM. Another way of saying this is that DCVax-l is likely to be effective against mesenchymal and classical GBM subtypes, but not so much proneural GBM. As luck would have it, the world changed the definition of GBM to only include mesenchymal and classical GBM, so the need to rely upon subtype analysis is probably no longer nearly as relevant.
Linda Liau submitted her comments.
BUT those comments were NOT hers. Instead someone posted those comments in her name, while they were actually taken from a Bill Malloy interview. So since she in fact did NOT send in the comments herself, one can not actually use that in assuming anything with regards to the guidance.
The publishing of the guidance has been awaited for a long time now.
Some argues, that the FDA will accept the endpoint changes. Below are some of these:
Comments from Sentiment Stocks:
Externally controlled trials differ in several important ways from the other trial designs identified in 21 CFR 314.126. Most notably, random assignment is not a feature of external control designs. As a result, there may be differences in patient characteristics or concomitant treatments in the trial population compared to the external control population that lead to differences in outcomes that are unrelated to the investigational treatment. In addition, the lack of blinding could introduce bias. For these reasons, external control designs are usually reserved for specific circumstances, such as trials of diseases with high and predictable mortality or progressive morbidity (e.g., certain malignancies or certain rare diseases) and trials in which the effect of the drug is self-evident (e.g., general anesthetics).
Despite the limitations of externally controlled trials compared with concurrently controlled trials, strong support for effectiveness can emerge from externally controlled trials, especially when
(1) the natural history of a disease is well defined
(2) the external control population is very similar to that of the treatment group
(3) concomitant treatments that affect the primary endpoint are not substantially different between the external control population and the trial population
(4) the results provide compelling evidence of a change in the established progression of disease.
Such results could include partial or complete response in a disease where spontaneous regression is not observed, or stabilization or improvement in function in a disease where progressive functional decline is well documented to occur over the duration of the treatment period in the trial. Another example of where there is strong evidence of drug effectiveness is reversal of clinical signs and symptoms following a toxic exposure or overdose after administration of a drug antidote. In all such circumstances, a detailed understanding of the full range of possible clinical outcomes, with a well-documented natural history of the disease in the absence of treatment, is essential to interpreting trial results and, therefore, drawing a conclusion about the effectiveness of the drug.
For example, compelling results may overcome challenges associated with less rigorous trial designs, such as those with an external control. As discussed above, a small externally controlled trial with an outcome markedly superior to the well-established natural history of a disease may provide a compelling case for drug effectiveness.
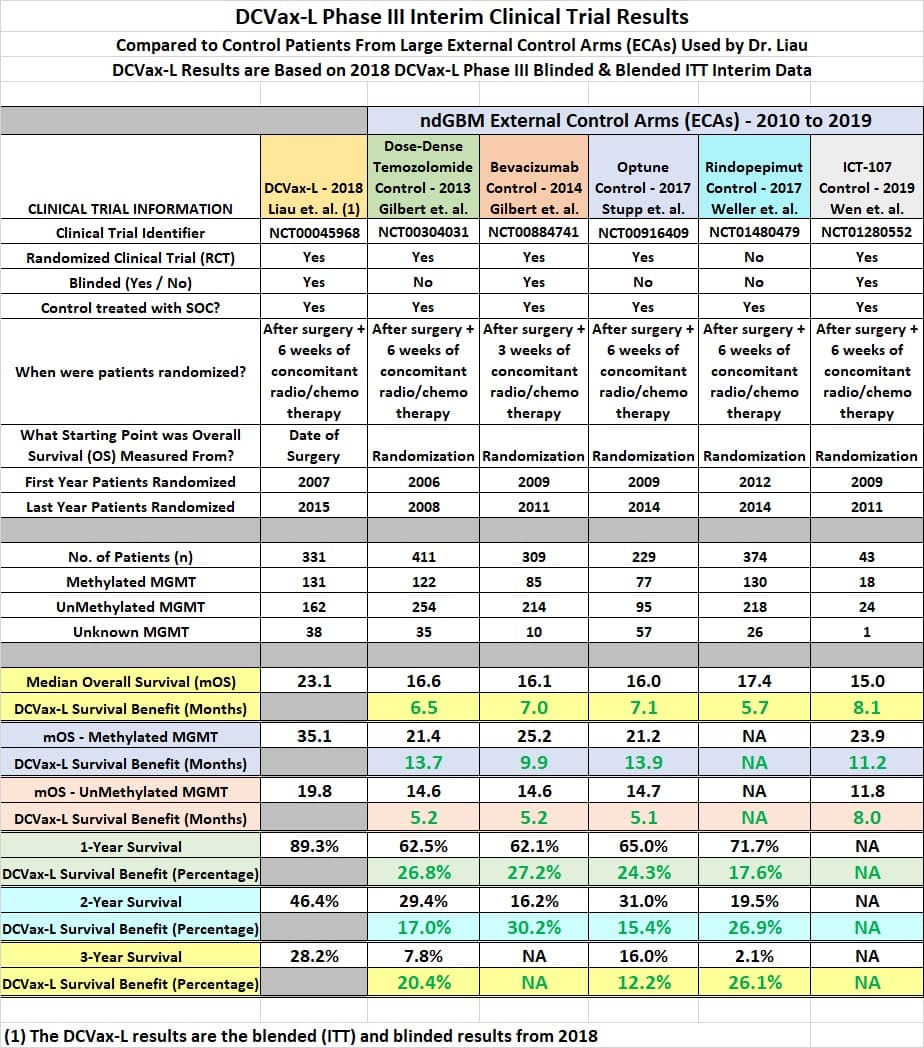
Until we all get wiser, enjoy the following overview of the DCVax-L interim clinical trial result.